
Jeder Mensch besitzt normalerweise 46 Chromosomen, die paarweise angeordnet sind; damit ist auch jedes Gen doppelt vorhanden: eines vom Vater und eines von der Mutter. Eine Ausnahme bilden lediglich die Gene auf den Geschlechtschromosomen oder Gonosomen: Bei einer Frau (Karyotyp 46, XX) sind auch diese Gene doppelt vorhanden, beim Mann (Karyotyp 46, XY) jeweils nur einmal auf dem X- und dem Y-Chromosom. Die Nicht-Geschlechtschromosomen werden auch als Autosomen bezeichnet und finden sich demzufolge bei weiblichen und männlichen Individuen gleichermaßen.
Im Normalfall sind die beiden Gene auf einem Paar von Autosomen gleich und führen zum Aufbau eines gleichen Proteins; in diesem Fall liegen die Gene im homozygoten Zustand vor (homós: griech. = gleich); man spricht von Homozygotie oder „Reinerbigkeit“. In einem anderen Fall kommt es vor, dass ein Gen auf dem einen Chromosom eine Veränderung erfahren hat, es ist mutiert, es liegt eine Mutation vor (mutare: lat. = sich verändern). Die Gene sind nicht mehr gleich und werden jetzt auch als Allele bezeichnet: Das nicht mutierte, normale oder Wildtyp-Allel führt unverändert zum Aufbau eines wirksamen Proteins. Das andere, mutierte Allel kann, je nach der Art der Mutation, zwar auch noch zum Aufbau eines Proteins führen; dieses Protein ist jedoch meist in seiner Funktion eingeschränkt.
In anderen Fällen wird gar kein Protein mehr gebildet, da die Mutation vorzeitig zum Abbruch des Proteinaufbaus geführt hat. Es liegen jetzt also mindestens zwei verschiedene Zustandsformen der Gene vor: das normale, unveränderte Wildtyp-Gen und das mutierte Gen mit einer bestimmten Mutation. In diesem Fall spricht man von „Allelen“ und meint damit zwei unterschiedliche Gene an demselben Ort eines Chromosomenpaares. Hierbei liegen die Gene (besser: Allele) also nicht im gleichen Zustand vor, sind also nicht homozygot, sondern sie liegen in einem unterschiedlichen und damit im heterozygoten Zustand vor (héteros: griech. = anders); man spricht von Heterozygotie.
Die Formen der Vererbung
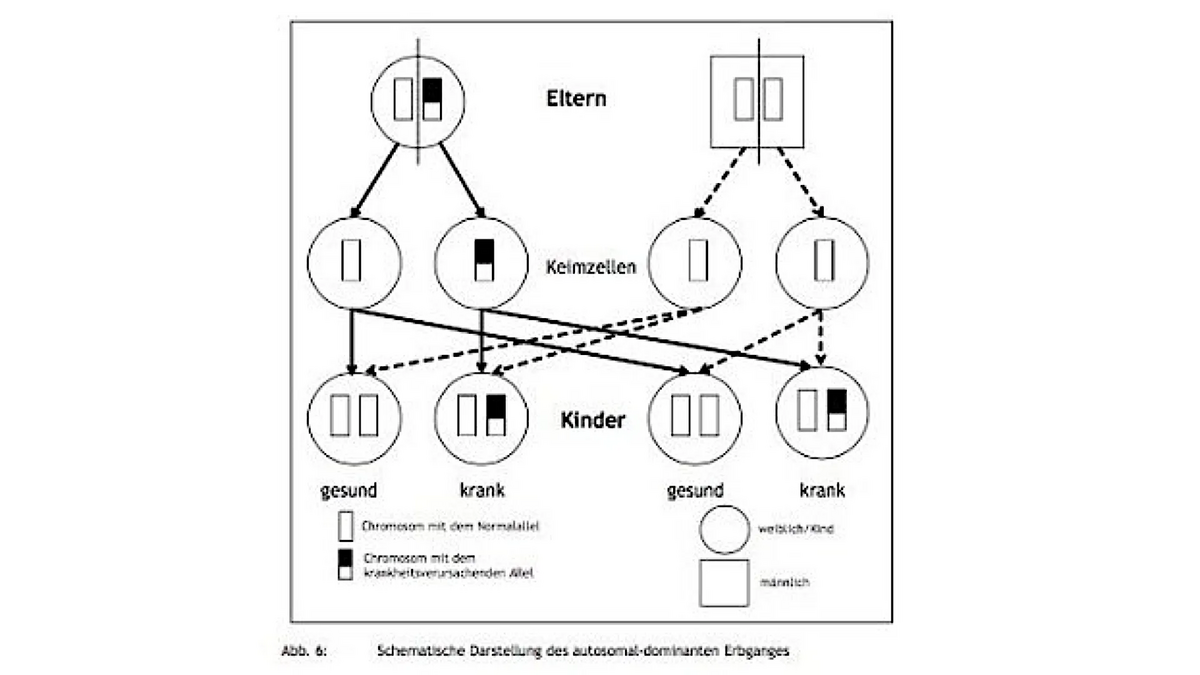

Hierbei handelt es sich um einen Erbgang, der aufgrund der Lokalisation der Gene auf einem Paar Autosomen unabhängig vom Geschlecht auftritt; es spielt also keine Rolle, ob es sich um eine weibliche oder männliche Person handelt. Nichts anderes besagt das Wort „autosomal“.
Von den beiden Genen auf einem Paar von Autosomen spielt, sofern sie durch eine genetische Veränderung (Mutation) nicht mehr identisch sind (man spricht jetzt von Allelen), meist das eine die entscheidende Rolle: Es dominiert (dominare: lat. = herrschen über, gebieten, befehlen); das andere Allel hingegen wird überspielt, überdeckt von dem dominierenden Allel: Es ist rezessiv (recedere: lat. = zurückweichen, entschwinden). Liegt Heterozygotie der Gene (Mischerbigkeit bei nicht identischen Allelen) vor und ist das mutierte Allel dominant gegenüber dem Normalallel (dieses ist dann rezessiv), wird sich das dominante Allel in seiner Wirkung ausprägen: Es kommt zum (gestörten) Aufbau eines Proteins, das in seiner Funktion eingeschränkt ist. Das rezessiv wirkende Allel kann diesen Ausfall des Proteins nicht kompensieren; als Folge kommt es zu einer Störung z. B. des Stoffwechsels der Nervenzelle und zur Ausprägung einer Krankheit, wie wir es von den dominanten Ataxieformen her kennen.
Bei dem autosomal-dominanten Erbgang reicht es also aus, dass lediglich eines der beiden Allele eines Chromosomenpaares eine Mutation erfahren hat (Krankheitsallel), damit eine Krankheit auftritt. Mischerbige (heterozygote) Träger eines dominanten Krankheitsallels, z. B. für die SCA1, werden also, eine entsprechende Lebensspanne vorausgesetzt, irgendwann im Laufe ihres Lebens eine Ataxie bekommen.
Da bei der Reifung der Geschlechtszellen (Ei- und Samenzellen) jeweils die Hälfte der Chromosomen in diese Geschlechtszellen gelangt (also nur 23 einzelne Chromosomen), können Ei- oder Samenzelle mit einer Wahrscheinlichkeit von 50 % wiederum das Krankheitsallel erhalten. Wenn diese Krankheitsallel-tragende Geschlechtszelle zur Befruchtung gelangt, kommt vom Partner meist ein Normalallel hinzu.
Aufgrund der Dominanz des Krankheitsallels führt es wieder zur Entstehung der Krankheit bei den Kindern. Nach dem Mendelschen Spaltungsverhältnis beträgt die Wahrscheinlichkeit für die Kinder eines Ataxie-Betroffenen, bei dem eine dominante Ataxie vorliegt, 50 %, wiederum Träger des Krankheitsallels zu sein. Diese Kinder werden als „Risikopersonen“ bezeichnet (aus dem Englischen: person at risk) und geben das Krankheitsallel, wiederum mit einer Wahrscheinlichkeit von 50 %, an ihre Kinder weiter, die ebenfalls wieder als Risikopersonen, diesmal jedoch mit einer 25 %igen Erkrankungswahrscheinlichkeit, bezeichnet werden.
Erkrankt ihr Elternteil an der dominanten Ataxie, steigt ihre eigene Erkrankungswahrscheinlichkeit auf 50 %; ist bei ihrem Elternteil mittels molekulargenetischer Methoden das Krankheitsallel nicht nachgewiesen worden, sinkt ihre eigene Wahrscheinlichkeit für diese Ataxie auf 0 %. Sie können also nicht von ihren Eltern erben, was diese nicht besitzen: in diesem Fall das Allel für eine dominante Ataxie. Da der Begriff „Risikoperson“ mehrdeutig ist und diskriminierend wirken kann, sollte besser von „Ataxie-gefährdeten Personen“ gesprochen werden.
Bei der Analyse eines Familienstammbaumes lässt sich im Idealfall nachweisen, dass in jeder Generation Ataxie-Betroffene auftreten und dass eine Ataxie nur dann wieder bei einem Nachkommen auftritt, wenn auch ein Elternteil erkrankt war oder ist.
Anders verhält es sich mit der Vererbung der Gene, die nicht auf einem Autosom, sondern auf einem Geschlechtschromosom (Gonosom) liegen.
Die uns hier interessierenden Gene, die seltene Ataxieformen (z. B. die geschlechtsabhängige, rezessive, spinozerebelläre Ataxie oder das Fragile X-Tremor-Ataxie-Syndrom) verursachen können, liegen auf dem X-Chromosom und zeigen eine rezessive Wirkung. Das heißt, dass bei einer weiblichen Person mit zwei X-Chromosomen das Krankheitsallel gewöhnlich nicht wirksam wird.
Während der Reifung der Eizellen werden auch die beiden X-Chromosomen getrennt und die gereifte, zur Befruchtung verfügbare Eizelle enthält damit nur ein X-Chromosom. Die Samenzelle des Partners, die entweder ein X- oder ein Y-Chromosom enthalten kann, bestimmt über das Geschlecht des gezeugten Kindes und somit auch darüber, ob es wiederum erkranken wird.
Enthält die Samenzelle ein X-Chromosom, wird ein Mädchen entstehen. Dieses Mädchen wird gesund sein, hat jedoch mit einer Wahrscheinlichkeit von 50 % entweder das X-Chromosom mit dem Krankheitsallel oder dem Normalallel von seiner Mutter geerbt.
Jede Tochter einer Frau, die ein rezessives Krankheitsallel auf einem X-Chromosom trägt, hat also eine Wahrscheinlichkeit von 50 %, ebenfalls wieder (Über-)Trägerin des Krankheitsallels zu sein. Diese Frauen werden als Konduktorinnen bezeichnet (conductor: lat. = Mieter, Pächter; conducere: lat. = übernehmen, beitragen). Enthält die Samenzelle ein Y-Chromosom, wird ein Knabe entstehen. Dieser Knabe hat (wie ein Mädchen auch) eine Wahrscheinlichkeit von 50 %, entweder das Krankheitsallel oder das Normalallel auf dem X-Chromosom von seiner Mutter zu erben.
Enthielt die befruchtete Eizelle das Krankheitsallel, wird der Knabe an der Ataxie erkranken: Er hat nur ein X-Chromosom; das andere Geschlechtschromosom ist ja ein Y-Chromosom (sonst wäre er nicht männlich!).
Bekommt er von der Mutter das Normalgen, bleibt er gesund. Söhne von Konduktorinnen haben also eine Wahrscheinlichkeit von 50 %, Genträger zu sein und damit zu erkranken, oder von 50 %, Nicht-Genträger zu sein und gesund zu bleiben. Alle Töchter eines Mannes, bei dem eine X-chromosomale Ataxie vorliegt, müssen gesunde Konduktorinnen sein, da sie vom Vater nur das X-Chromosom mit dem Krankheitsallel bekommen können. Alle Söhne dieses Mannes sind gesund und Nicht-Genträger für das Krankheitsallel, da sie von ihrem Vater ja das Y-Chromosom bekommen.
Bei der Stammbaumanalyse sieht es manchmal so aus, als ob die Ataxie vom kranken Großvater über dessen gesunde Töchter auf die wiederum erkrankten Enkelsöhne „springt“, also in jeder zweiten Generation auftritt.
In Wirklichkeit ist das Krankheitsallel in jeder Generation vorhanden, führt jedoch bei den weiblichen Familienmitgliedern, da sie zwei X-Chromosomen haben, und wegen der rezessiven Wirkung nicht zu der Ataxie.
Die mitochondriale Vererbung stellt einen Sonderfall dar und unterliegt nicht den Mendelschen Erbregeln. Als Mitochondrien werden Zellorganellen bezeichnet, die besonders für die Bereitstellung der Energie verantwortlich und an den Atmungsprozessen der Zelle beteiligt sind. Auch die Mitochondrien enthalten DNS; diese wird jedoch nach anderen Gesetzmäßigkeiten vererbt.
Mitochondrien können sich, je nach dem Energiebedarf der Zelle, unabhängig von dieser vermehren und garantieren so eine optimale Energiebereitstellung, je nach den Anforderungen. So enthalten die Muskelzellen bedeutend mehr Mitochondrien als Zellen des Fettgewebes. Auch in der DNS eines Mitochondriums kann es zu Mutationen kommen, die zu einem gestörten Aufbau eines Proteins, das z. B. an der Zellatmung beteiligt ist, führt.
Eine Mutation in einem einzelnen Mitochondrium unter vielen in einer Zelle wird sich nicht als Krankheit auswirken; jedoch kann sich dieses Mitochondrium in der Zelle vermehren und auch bei der Zellteilung in die Tochterzelle gelangen und somit ein ganzes Zellgewebe beeinflussen. Dies kann schon Auswirkungen auf den Stoffwechsel des Organs haben. Das Nebeneinandervorkommen von normalen und „mutierten“ Mitochondrien wird als Heteroplasmie bezeichnet.
Mitochondrien werden nur über die mütterliche Linie vererbt. Die zur Befruchtung gelangende Eizelle stellt allein die Mitochondrien und damit die Energie für den sich entwickelnden Embryo bereit. Die Samenzelle stellt lediglich den Zellkern mit der dort enthaltenen DNS für den Embryo bereit. Das hat für die Vererbung die Bedeutung, dass ein an einer mitochondrialen Ataxie erkrankter Mann seine mitochondriale Mutation nicht an seine Kinder weitergeben kann, diese also gesund bleiben. Lediglich bei den Kindern einer an einer mitochondrialen Ataxie erkrankten Frau kann es zum Wiederauftreten der Ataxie-Krankheit kommen. Da die Anzahl der mutierten Mitochondrien in den Zellen den Schweregrad der Krankheit bestimmt, ist nicht vorauszusagen, mit welcher Wahrscheinlichkeit die Ataxie-Erkrankung wieder auftreten wird.Theoretisch liegt diese Wahrscheinlichkeit zwischen 0 % (das Kind bekommt kein mutiertes Mitochondrium von der Mutter) und 100 % (alle vererbten Mitochondrien tragen eine DNS-Mutation).